引言:
就单种罕见病,由于患病人群少、市场需求少、研发成本高,很少有制药企业等关注其治疗药物的研发,因此这些罕见病患者的用药被形象地称为“孤儿药”。虽然患者群体少,但每一位罕见病病友对孤儿药的需求却是百分百的,并且整个罕见病的患病群体是十分庞大的,据不完全统计,在中国有超过千万的罕见病患者。而在国内,受罕见病政策缺失、市场不成熟、研发技术欠缺、社会关注较低等因素的制约,罕见病药物市场研发几乎为空白。然而近年来随着国家政府层面对罕见病领域的关注与支持,这一困境,或许在不久的将来会有所突破。
在今年2月初国务院办公厅印发《关于进一步改革完善药品生产流通使用政策的若干意见》(以下简称《意见》)。《意见》中强调到要借鉴国际先进经验,探索按罕见病、儿童、老年人、急(抢)救用药及中医药(经典方)等分类审评审批,保障儿童、老年人等人群和重大疾病防治用药需求。
而今年“两会”期间,更多的委员或代表为罕见病提案,呼吁加大对罕见病或孤儿药研发的支持力度,及时出台相关的鼓励措施。至5月11日国家药监总局(CFDA)发布《关于鼓励药品医疗器械创新加快新药医疗器械上市审评审批的相关政策》的征求意见稿,其中明确指出“支持罕见病治疗药物和医疗器械研发。由卫生计生部门公布罕见病目录,建立罕见病患者注册登记制度。罕见病治疗药物和医疗器械申请人可提出减免临床试验申请,加快罕见病用药医疗器械审评审批。对于国外已批准上市的罕见病治疗药物和医疗器械,可有条件批准上市,上市后在规定时间内补做相关研究。”
6月2日,国家科技管理信息系统公共服务平台官网发布了“关于国家重点研发计划‘精准医学研究’和‘生殖健康及重大出生缺陷防控研究’重点专项2017年度项目安排公示的通知”,其中在“精准医学研究”重点项目专项拟立项中“中国重大疾病与罕见病临床与生命组学数据库”(序号19),中央财政经费达4977万元。
整体而言,国内政府部门已经在不断加大了对罕见病或孤儿药研发的关注与支持,社会层面的关注力度也在不断增强。未来相信会有更多的惊喜,让我们一起拭目以待!
目前,在孤儿药研发领域,美国仍占据着主要地位,其次是欧盟等。以美国为例,到今日为止(7月9日),自1983年《孤儿药法案》实施以来,美国FDA已批准上市的孤儿药已达到625项,而获得孤儿药资格的近4200项。而在2017年上年(至6月底),FDA共批准孤儿药27个(按药物名称,如按疾病对应则有35项),而授予孤儿药资格235项。这些数据也远远超过欧盟等。
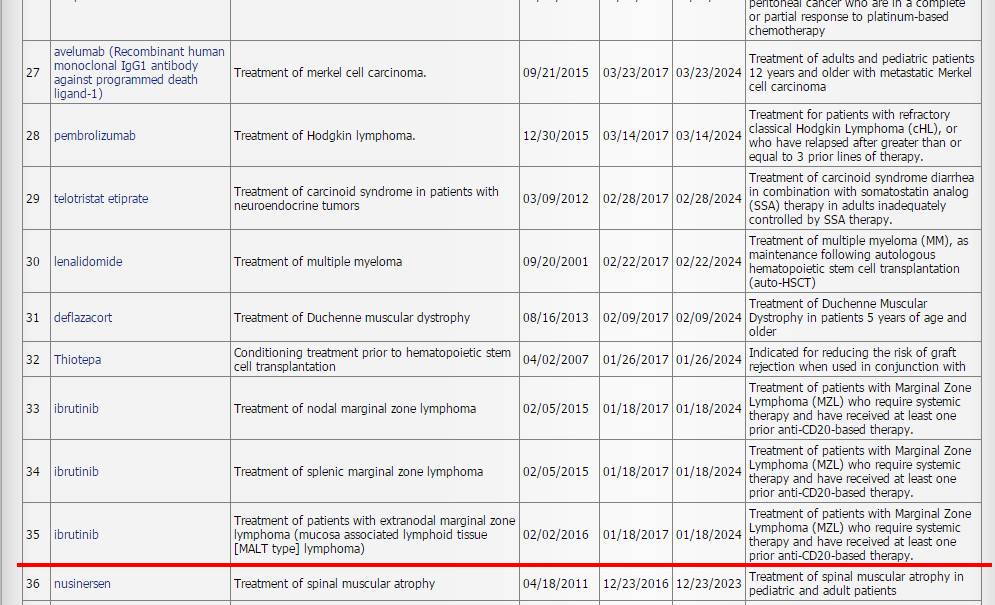
图片来源:美国FDA官网—Search Orphan Drug Designations and Approvals
相关孤儿药研发或审批信息盘点
以下主要盘点国外已获得批准上市的孤儿药
(或个别孤儿药地位),排序不分先后:
1.美国FDA批准首个治疗杜氏肌营养不良(DMD)的类固醇药物Emflaza

美国食品和药物管理局(FDA)已批准Marathon Pharma公司的药物Emflaza(deflazacort),用于5岁及以上杜氏肌营养不良(DMD)患者的治疗。此次批准,使Emflaza成为全球获批治疗DMD的首个皮质类固醇药物,同时也是全球获批治疗DMD的第二款药物。此前,FDA已授予Emflaza孤儿药地位和优先审查资格。
去年9月底,FDA有条件批准了治疗DMD的首个药物Exondys 51(eteplirsen),该药来自Sarepta公司,专门适用于抗肌营养不良蛋白基因(dystrophin gene)中存在确证突变可导致51号外显子跳跃(exon 51 skipping)的DMD患者;据估计,该类患者约占全部DMD患者的13%。Exondys 51可解决导致该类DMD病例的根本病因,能够产生有功能的抗肌营养不良蛋白,在临床研究中具有广泛良好的安全性、耐受性和疗效。
2.FDA授予EB-101孤儿药地位,用于治疗大疱性表皮松解症(EB)

5月,Abeona公司宣布FDA已授予EB-101基因治疗孤儿药资格用以治疗营养不良性大疱性表皮松解症(DEB),包括隐性营养不良性大疱性表皮松解症(RDEB),这是一种严重威胁生命的遗传性皮肤病,主要特征是全身皮肤水疱、糜烂等。而早在今年3月初,Abeona 也已宣布欧洲药品管理机构(EMA)孤儿药品委员会授予了EB-101用于治疗隐性营养不良性大疱性表皮松解(RDEB)的孤儿药地位。
EB-101是一种自体同源、体外转移(ex-vivo)的基因疗法,COL7A1基因被导入自体同源角质细胞以治疗RDEB。1期/ 2期的临床试验正在进行的2期部分(nct01263379)。EB-101最近的临床数据是由该公司的合作者斯坦福大学在皮肤病学会(SID)会议中提出的,并且基于这些临床和药后两年多的跟进,证明了EB-101治疗效果明显,伤口均愈合> 50%。
3. SMA药物SPINRAZA® (Nusinersen)获欧盟批准,成欧洲首个脊髓性肌萎缩症(SMA)治疗药物
近期,欧盟委员会(The EuropeanCommission ,简称EC)已批准SPINRAZA(nusinersen)用于治疗5q型脊髓性肌萎缩症(5q spinalmuscular atrophy),这也是欧洲首个批准的脊髓性肌萎缩症(SMA)治疗药物。SMA可分为多种类型,也有5q-SMA与非5q-SMA之分,而5q-SMA是SMA中最常见的类型(1-4型),约占SMA病例中的95%,是由5号染色体上的SMN1(运动神经元生存蛋白1)基因突变所引起的,因此被称为5q型SMA。
在16年10月,欧洲药物管理局(EMA)已通过了SPINRAZA 作为脊髓性肌萎缩疗法的上市许可申请,欧洲药物管理局人用药品委员会(CHMP)将SPINRAZA的申请列为“加速评估”。同时,百健也已在日本、加拿大和澳大利亚提交监管申报文件。
在去年12月,美国FDA率先批准了SPINRAZA上市,使得SPINRAZA成为首个也是唯一在美国获批治疗SMA的药物。而在今年5月FDA又授予Cytokinetics的实验性药物CK-2127107治疗SMA的孤儿药地位(orphan drug designation)。
CK-2127107由Cytokinetics与日本药企安斯泰来(Astellas)合作开发,这是一种新一代快速骨骼肌肌钙蛋白激活剂(FSTA),开发用于脊髓性肌萎缩症(SMA)、慢性阻塞性肺病(COPD)及其他一些与骨骼肌无力和/或疲劳相关疾病的潜在治疗。目前,该公司正在探索该药改善SMA患者肌肉功能和身体活动能力的潜力,一项正在开展的 II 期临床研究数据预计将在今年晚些时候获得。
4. FDA批准Radicava(edaravone)依达拉奉用于ALS治疗
5月5日(当地),美国FDA宣布批准了Radicava(edaravone)用于治疗肌萎缩侧索硬化(ALS),这也是美国FDA22年来批准的首款ALS新疗法,这种药物必须静脉输注。
依达拉奉治疗ALS的疗效在日本进行了为期6个月的临床试验。在试验中,137名参与者随机接受依达拉奉或安慰剂治疗。在第24周的观察中,与接受安慰剂的人相比,接受依达拉奉的患者的日常功能的临床评估指标下降较少。而另一个药物riluole是在1995年批准的。
5. FDA批准首个针对晚发婴儿型神经元蜡样脂褐质沉积症(CLN2)的疗法
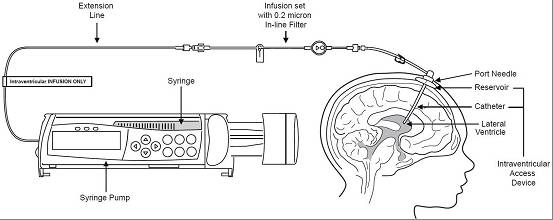
脑室内输液系统的设置(来源:FDA Approved Drug Products,Products on BLA761052)
4月27日(当地时间),美国FDA批准了Brineura(cerliponase alfa),成为首个针对晚发婴儿型神经元蜡样脂褐质沉积症(CLN2)的疗法,用于缓解3岁和3岁以上的CLN2患者行走能力(步行)的丧失, CLN2又被称为三肽基肽酶-1(TPP1)缺乏症,是Batten病的一种。这也将是FDA已批准的最昂贵的疗法之一,每年花费总计702000美元。
Brineura是一种酶替代疗法,它的活性成分(cerliponasealfa)是人类TPP1的重组形式,这正是CLN2患者所需要的。3岁和3岁以上的儿科患者,每隔一周通过脑室内输注一次,推荐剂量为300mg,之后进行电解质输注。输注Brineura的完整过程,包括了所需的脑室内电解质输注,持续约4.5个小时。建议在开始输液前30-60分钟,先施用抗组胺药物或皮质类固醇。在临床试验中,Brineura的疗效得到了验证,但尚未建立3岁以下患者使用Brineura的安全性和有效性。同时,FDA也公布了一些常见的不良反应,比如发烧、呕吐等。
6.阿斯利康神经脊髓炎新药inebilizumab获欧盟委员会授予孤儿药资格
阿斯利康神经药物产品管线近日在欧盟监管方面收获好消息,其药物inebilizumab(前身为MEDI-551)被欧盟委员会(EMA)授予孤儿药资格,用于治疗视神经脊髓炎谱系疾病,这将为该药物在未来的研发和上市提供有利的政策支持。
Inebilizumab最初是由MedImmune研发的人源化单克隆抗体,与靶点CD19具有高亲和力,CD19为B细胞表面抗原,在B细胞的发育过程中的各个阶段均有表达,并且在浆细胞表面亦有表达。视神经脊髓炎的发病率约为十万分之五,该疾病会导致严重的肌无力和麻痹、视力丧失、呼吸衰竭、肠道和膀胱功能障碍和神经性疼痛,目前还没有治疗该疾病的有效手段。
7. FDA批准AUSTEDO™(deutetrabenazine)用于治疗与亨廷顿舞蹈症相关的“舞蹈病症状”

4月3日,FDA(Food andDrug Administration,简称FDA)批准了用于治疗“舞蹈病症状”(与亨廷顿舞蹈症相关)的药物AUSTEDO™(deutetrabenazine;SD-809),AUSTEDO™是FDA批准的首个氘代产品,也是第二个获得FDA批准的针对亨廷顿舞蹈症(Huntington’s disease ,简称HD)的药物,该药物先前FDA已授予孤儿药资格。
AUSTEDO™旧称为SD-809,2016年5月Teva收到FDA“完全回应函”(CompleteResponse Letter),并要求Teva检查血液中某些代谢物的浓度水平,药物的批准也被延迟。但FDA并没有要求新增临床试验。
AUSTEDO™是一种单胺囊泡转运体2(VMAT2)的口服小分子抑制剂,FDA的批准是基于随机、双盲、安慰剂对照的多中心第三期临床试验的研究结果,减轻HD患者“舞蹈病”的疗效很明显。
据相关分析人士预测,到2023年AUSTEDO™的销售额将达到8.5亿美元
8. FDA批准首个治疗原发进展型多发性硬化症的新药(OCREVUS™)

3月底,美国食品药品监督管理局(food and drug administration,简称FDA)批准了OCREVUS™(ocrelizumab)用于治疗复发-缓解型多发性硬化(RRMS)和原发进展型多发性硬化(PPMS),这是FDA批准的首个针对PPMS的药物。
OCREVUS™(ocrelizumab)是一种针对CD-20阳性B细胞的人源化单克隆抗体。这是一种静脉输注药物,患者每年需注射两次,每次600 mg,每年费用约65000美元。OCREVUS的上市许可申请(MAA)也已获得欧洲药品管理局(EMA)的批准,目前正在审核流程中。
到目前为止,FDA已经批准了13种用于治疗复发性MS的药物(1993年首个),但是却没有一种可以治疗原发进展型多发性硬化(PPMS),OCREVUS是第一个。另外,OCREVUS是一种用于治疗成人患者复发或原发进行型多发性硬化的药物,对儿童是否安全、有效还未知。
9. FDA授予tavokinogene telsaplasmid治疗不可切除性转移性黑色素瘤的孤儿药资格
oncoSec Medical公司已宣布美国食品和药物管理局已授予tavokinogene telsaplasmid(pIL-12,一种可表达白细胞介素-12[IL-12]的质粒)治疗不可切除性转移性黑色素瘤的孤儿药资格。
IL-12是一种体内自然产生的蛋白质,具有免疫刺激功能。pIL-12是OncoSec先导候选产品ImmunoPulseIL-12中的活性生物制剂,后者是一种潜在首创(first-in-class)的瘤内抗癌基因疗法,通过瘤内注射及短脉冲电穿孔递送至肿瘤微环境中,产生受控的、局部表达的IL-12,从而使免疫系统能够靶向并攻击肿瘤细胞,其基于OncoSec公司的核心技术平台Immuno Pulse开发。
10. FDA批准首个皮下注射用C1酯酶抑制剂用于遗传性血管性水肿
6月22日,美国FDA批准CSL Behring公司的Haegarda用于青少年及成年患者遗传性血管性水肿(HAE)发作预防,这是FDA批准用于该疾病的首个皮下注射用C1酯酶抑制剂(人类)。经过正规培训后,皮下注射方式可令患者或看护者在家自行注射变得更容易。HAE患者的C1酯酶抑制剂缺失或水平低,在某些病例中,C1-INH水平是足够的,但这种蛋白不能发挥正常的功能。C1-INH缺陷在人的基因序列中,这也是其是一种遗传性疾病的原因。
HAE因没有充分数量的血浆蛋白C1酯酶抑制剂(或C1-INH)而引起。没有充分数量可以工作的C1-INH,体内的血管及毛细血管可以产生泄露,使得液体在周边部位积聚。
11. 罗氏片剂剂型Esbriet(比非尼酮)获欧盟批准治疗轻度至中度特发性肺纤维化(IPF)
瑞士制药巨头罗氏(Roche)近日宣布,欧盟委员会(EC)已批准新的速释片剂剂型Esbriet(pirfenidone,比非尼酮)用于轻度至中度特发性肺纤维化(IPF)患者的治疗。罗氏已计划于2017年在数个欧洲国家推出片剂Esbriet。
IPF是一种不可逆转的、致命性、渐进性肺部疤痕。相关临床数据显示,Esbriet能够减缓IPF的进展。与安慰剂相比,Esbriet治疗52周使IPF患者的死亡风险显著降低48%(p=0.01)。在治疗的第52周时预测的FVC从基线降低幅度≥10%的患者比例,Esbriet治疗组为17%,安慰剂组为32%。此外,与安慰剂相比,Esbriet显著降低了6分钟步行测试距离的下降。
在美国,Esbriet胶囊剂型于2014年10月获得美国FDA批准上市,目前该药已在全球多个国家上市销售。2017年初,Esbriet 801mg和267mg片剂获得了FDA批准,作为治疗IPF的新治疗选择。其中801mg片剂,可作为一种维持治疗选择,减少IPF患者每日服药数量。
在美国和欧盟,Esbriet均被授予了治疗IPF的孤儿药地位。
注:本文内容仅供参考、交流,不具有任何商业用途的指导性建议。由于受到专业、资料搜集等因素的局限性,难免对一些药物资讯有所遗漏,敬请理解。
欢迎投稿邮箱:info@cord.org.cn
资讯 | 数据 | 孤儿药 | 会议 | 政策 | 患者组织
——罕见病行业门户信息平台——
www.raredisease.cn
-----------------------------------------