类风湿关节炎(Rheumatoid arthritis, RA)是一种以慢性侵蚀性关节病变为主要表现的全身性自身免疫性疾病[1]。我国类风湿关节炎患病率一直保持在 0.2% ~ 0.4% [2],并且患病率随年龄增加而显著提升[3]。随着病情进展,患者逐渐出现滑膜炎症,软骨和骨质的破坏,最终导致关节畸形[2]。RA 致残率较高,但是通过规范化治疗,可以达到短期内控制炎症,进而改善关节功能、控制疾病进展的治疗目标[2]。
RA 的治疗在过去的一百多年间经历了很多的探索,从传统的 DMARDs (disease-modifying anti-rheumatic drugs),到以靶向细胞外单一炎症因子的大分子生物制剂,如今已进入新兴的 Janus 激酶(Janus kinase, JAK) 通路小分子抑制剂靶向治疗时代[1, 4, 5]。传统 DMARDs 虽可缓解关节肿胀和疼痛,但起效慢、药理机制尚不明确,并会引起不同程度的不良反应[2, 4]。用于 RA 治疗的生物制剂包括 TNF 拮抗剂、利妥昔单抗(rituximab)、托珠单抗(tocilizumab)等,虽起效迅速,但易引起细菌、真菌和病毒感染,并使淋巴瘤患病风险增加[4]。新一代 RA 治疗药物以抑制 JAK 为靶点,已经被用于治疗 RA 以及其他炎性疾病[5]。
JAK激酶作为药物靶点在RA中的作用
RA 是一类以病变滑膜中活化免疫细胞浸润及促炎症细胞因子、促基质降解酶类被大量释放为特征的自身免疫性疾病,主要表现为滑膜炎及软骨与骨破坏,从而导致持续性关节损伤。其中具体发病机制如图所示:首先滑膜炎症组织释放的促炎症细胞因子及趋化因子,可招募 B 细胞、 T 细胞、巨噬细胞等多种免疫细胞浸润至关节局;并且通过 JAK 通路在内的多种信号传导通路促使免疫细胞活化及产生更多的促炎症细胞因子;被大量释放至局部的促炎症因子募集并活化更多的免疫细胞,从而形成一个慢性的炎症级联放大反应,最终导致持续性关节损伤。因此,促炎症细胞因子及其细胞内信号传导通路在 RA 复杂的调控网络中扮演关键角色[1]。

图1. JAK 途径在 RA 慢性周期性炎症演变中的作用
RA 发病过程中的关键细胞因子包括以下 I/II 型细胞因子家族成员:干扰素(IFN)α,IFNβ; 白介素(IL)-1,IL-6,IL-7,IL-10,IL-12,IL-15,IL-17,IL-18,IL-21,IL-23; 转化生长因子(TGF)-β; 和肿瘤坏死因子(TNF)[1]。生理情况下,这些被分泌至胞外的细胞因子或促进炎症,或抑制炎症,相互调节,最终维持机体的免疫稳态。然而, RA 患者体内的促炎/抗炎细胞因子稳态被打破,其疾病的发生发展与细胞因子之间的相互调控失衡相关[5]。
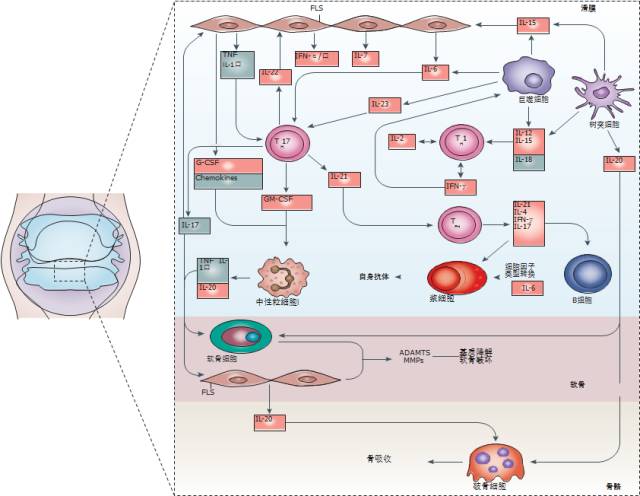
图2.RA 发病机制中的细胞因子网络
JAK-STAT 通路是细胞外促炎症细胞因子通过膜受体向核内传导炎性信号的重要信号通路。JAK 是英文 Janus kinase 的缩写。Janus 在罗马神话中是掌管开始和终结的两面神。之所以称为两面神激酶,是因为 JAK 既能磷酸化与其相结合的细胞因子受体,又能磷酸化转录因子 STATs 。细胞因子与细胞膜表面的相应受体结合后引起受体分子二聚化,这使得与受体偶联的 JAK 相互接近,并通过交互的酪氨酸磷酸化作用而活化。JAK 激活后催化受体上的酪氨酸残基发生磷酸化修饰,继而这些磷酸化的酪氨酸位点与周围的氨基酸序列形成「停泊位点」(docking site),将 STAT 蛋白招募到这个「停泊位点」。最后,激酶 JAK 催化结合在受体上的 STAT 蛋白发生磷酸化修饰,活化的 STAT 蛋白以二聚体的形式进入细胞核内与靶基因结合,诱导大量炎症相关基因的转录[6]。
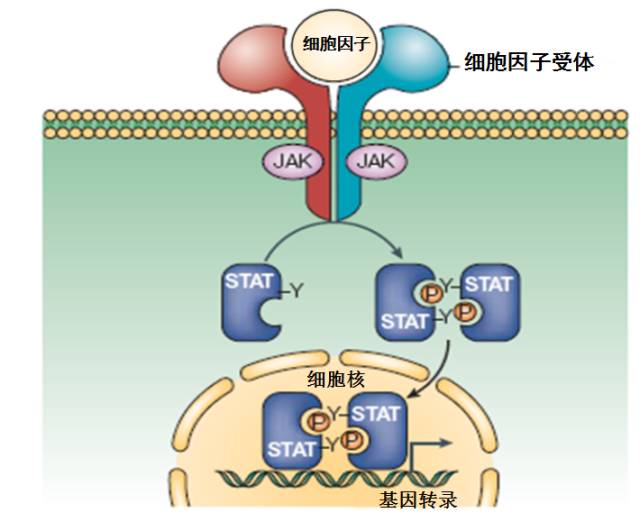
图3. JAK-STAT 信号通路三部曲
值得一提的是,不同的细胞因子通过 JAK 激或其相应的 STATs,通过不同的 STATs 对实现不同基因的开启模式[1]。
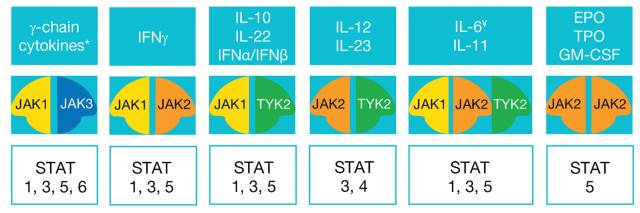
图4 细胞因子通过 JAK 与下游 STATs 之间存在特异的、多样化的配对关系
新型小分子 JAK 抑制剂通过模拟 ATP 的结构(但无磷酸基团)竞争性抑制 JAK 激酶的磷酸化,从而阻断细胞因子受体对 STAT 的募集及磷酸化,导致 STAT 不能活化并无法入核启动炎症相关基因的转录[1]。
作为一种靶向 JAK 激酶的小分子抑制剂,托法替布是 RA 治疗中的一种创新和进步。
靶向JAK的口服小分子抑制剂——托法替布
JAK 家族共包括四个非受体酪氨酸激酶成员,即 JAK1、JAK2、JAK3 和 TYK2[6]。研究表明大多数激酶均含有一个蛋白序列和结构高度保守的 ATP 结合结构域,其可催化 ATP 中的磷酸基团转移至特定靶蛋白,使蛋白发生磷酸化并活化。托法替布则被设计为一种与 ATP 高度相似、但无磷酸基团的小分子化合物[1],这种结构相似性使其能与 ATP 竞争性结合并抑制 JAK 激酶活性。但是我们如何才能找到一种对 JAK 激酶具有高度选择性,而对其他激酶无有效抑制的小分子化合物呢?通过对化合物进行侧链修饰等药物化学手段,筛选后得到了托法替布。
但托法替布对于不同 JAK 家族成员的抑制程度略有不同,对 JAK1(IC50=3.2nM)和 JAK3(IC50=1.6nM)的抑制效率明显高于 JAK2(IC50=4.1nM),而对 TYK2 的抑制效率最低。我们都知道通常 JAK 通常以同源/异源二聚体形式传导活化信号,研究表明托法替布对 JAK1/JAK3 组合形式的二聚体存在有高度选择的抑制效应(IC50=56nM),JAK1/JAK2(IC50=287nM)次之,但是对 JAK2/JAK2 组合的抑制效应较弱(IC50=1377nM)[1]。

图5. 通过 JAK / STAT 组合的 RA 和细胞因子信号传导的发病机制中的关键细胞因子
另外,托法替布通过抑制细胞内 JAK 信号通路传导,抑制免疫细胞活化及促炎症细胞因子的释放,直接或间接抑制了包括 IL-6、TNF-a 等多个促炎症细胞因子的产生及效应,阻断炎症的级联放大反应,使 RA 患者体内促炎症细胞因子水平呈指数级下降,从而达到治疗 RA 的目的[1]。

图6. 托法替布的作用机理
托法替布的临床价值
目前,RA 治疗中作为金标准的 DMARDs(包括甲氨喋呤)对于 RA 的治疗停药率高,导致治疗有效率低于 60%;而在接受生物制剂治疗的患者中仍有 28%~41% 的患者疗效不佳[7, 8] 。这部分患者亟需更有效的药物治疗。
托法替布可与柠檬酸与形成稳定的柠檬酸盐结晶,该特性使其适用于口服给药。尚杰®是目前美国 FDA 和国家食品药品监督管理局(CFDA)唯一获批用于治疗RA的口服小分子靶向 JAK 激酶抑制剂。托法替布用于 RA 治疗的推荐用量为每次 5 mg,每日两次(BID),可与非生物类 DMARDs 组合使用[9,10]。口服 Tofacitinib 之后,1 小时就可以达到血药浓度高峰,半衰期为 3 个小时,经过 12 个小时的代谢,基本上达到药物浓度的低谷[1]。托法替布可经过肝肾双通道代谢,其中肝脏代谢占 70% ,肾脏代谢占 30%[11]。

图7. 口服托法替布(BID)的血浆药物浓度波动情况
口服托法替布用药安全、便捷、经济。一项长期安全性研究结果证实,包括严重感染、机会感染、结核、肿瘤、心血管事件、消化道穿孔等安全性事件长期稳定在较低发生率[12]。一项荟萃分析结果证实:托法替布治疗期间的严重感染与恶性肿瘤发生率与生物制剂类似[13]。
小结
托法替布是一种小分子靶向药物,主要通过抑制胞内 JAK 依赖的细胞因子信号通路来发挥 RA 治疗作用。与生物制剂类 DMARDs 靶向拮抗单一细胞因子不同,托法替布靶向抑制炎症因子信号传导通路中的关键成分 JAK 激酶,进而控制炎症反应水平并调节机体免疫系统回归稳态。综上所述,口服小分子靶向药物托法替布的上市将有效助力 RA 治疗,为广大患者带来新的选择。
参考文献
[1]Hodge J A, Kawabata T T, Krishnaswami S, et al. The mechanism of action of tofacitinib - an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis.[J]. Clinical & Experimental Rheumatology, 2016, 34(2):318.
[2] 中华医学会风湿病学分会. 类风湿关节炎诊断及治疗指南 [J]. 中华风湿病学杂志, 2010, 14(4):265-270.
[3]Li R, Sun J, Ren L M, et al. Epidemiology of eight common rheumatic diseases in China: a large-scale cross-sectional survey in Beijing[J]. Rheumatology, 2012, 51(4):721.
[4]Scott D L, Wolfe F, Huizinga T W. Rheumatoid arthritis[J]. Sykepleien Fag, 2010, 376(9746):1094.
[5]Schwartz D M, Bonelli M, Gadina M, et al. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases[J]. Nature Reviews Rheumatology, 2016, 12(1):25-36.
[6]Shuai K, Liu B. Regulation of JAK–STAT signaling in the immune system[J]. Nature Reviews Immunology, 2003, 3(11):900-911.[7]Bernatsky S, Feldman D E. Discontinuation of Methotrexate Therapy in Older Patients with Newly Diagnosed Rheumatoid Arthritis[J]. Drugs Aging, 2008, 25(10):879-884.
[8]Agarwal S K, Glass R J, Shadick N A, et al. Predictors of discontinuation of tumor necrosis factor inhibitors in patients with rheumatoid arthritis[J]. J Rheumatol, 2008, 35(9):1737-1744.
[9]van Vollenhoven RF, Fleischmann R, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis[J]. N Engl J Med. 2012, 367(6):508-519.
[10]Heijde D V D, Tanaka Y, Fleischmann R, et al. Tofacitinib (CP‐690,550) in patients with rheumatoid arthritis receiving methotrexate: Twelve‐month data from a twenty‐four–month phase III randomized radiographic study[J]. Arthritis & Rheumatology, 2013, 65(3):559-570.[11]Dowty M E, Lin J, Ryder T F, et al. The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a janus kinase inhibitor, in humans[J]. Drug Metabolism & Disposition the Biological Fate of Chemicals, 2014, 42(4):759-73.
[12]Cohen S B, Tanaka Y, Mariette X, et al. Long-term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: integrated analysis of data from the global clinical trials[J]. Annals of the Rheumatic Diseases, 2017:annrheumdis-2016-210457.
[13]Strand V, Ahadieh S, French J, et al. Systematic review and meta-analysis of serious infections with tofacitinib and biologic disease-modifying antirheumatic drug treatment in rheumatoid arthritis clinical trials[J]. Arthritis Research & Therapy, 2015, 17(1):1-9.